80%中国临床数据不真实,是信达生物被拒的“力证”还是“托词”?
2月10日晚间,FDA召开肿瘤药物咨询委员会ODAC,对信迪利单抗注射液的新药上市申请(BLA)审评问题进行讨论并投票。最终以14票反对1票赞成,要求信达生物补充额外的临床试验,证明其对美国患者有效,FDA拒绝批准信迪利单抗。
信达生物信迪利单抗此次作为国内创新药单一国家临床数据登陆美国市场的“试金石”,既代表着其对信达生物自身的考验,也可认为是代表着中国创新药物公开接受过堂考,获取国际认可的标志。
揭“黑历史”
80%中国临床数据不真实确是现状?
然而值得注意的是,在评审委员针对信迪利单抗提出多个争议点时,FDA直接引用中国药监部门的报告,称80%的中国临床研究存在欺诈或者不够标准。再次引起业界讨论,这确定是中国新药临床研究当下的真实情况?

细心的读者不难发现,FDA引用的报告数据为中国食品药品监督管理局2016年报告。这份报告的源头需要从中国药界“7.22”惨案说起。2015年7月22日中国国家食品药品监督管理总局(CFDA)发布中国药界研发史上最严数据核查要求,对1622个新药申请进行临床数据核查,其中80%的药物临床试验未能通过核查。
自此便开启了中国药物新政改革,化药注册分类改革,BE备案制、国外临床试验数据被认可,国际新药临床试验可以同步开展等政策已得以实施,建立专利链接制和数据保护制度、新药品注册管理办法修改……
经过多年政策组合拳与顶层设计,中国药物研发环境经过溯本清源,中国新药研发已基本实现与国际接轨。尤其是新药创制专项实施以来,重大品种研发取得累累硕果。BTK抑制剂、第3代EGFR抑制剂、PARP抑制剂、CDK4/6抑制剂等国产小分子靶向药陆续上市,以及现下极其内卷的PD-1/PD-L1,打破了进口靶向药对国内市场的垄断。
且中国药物研发速度与数据获得全球多个国家的支持。最近也是最有说服力的为中国研究的新冠疫苗,得到全世界的认可,彰显中国药物创新与国际接轨成果。
FDA引用“7.22”数据
“力证”还是“托词”?
此次ODAC审评信迪利单抗,引用“7.22”惨案报告数据,是FDA拒绝信迪利单抗以中国临床数据上市的有力证据,还是一种对中国临床数据不信任的借口?
笔者认为,FDA引用已经过时的数据,实际是缺乏说服力的,或者说不是成为信迪利单抗不被FDA批准的有力数据支撑证据。
追溯信迪利单抗药物研发历程,据药智全球上市药品筛选系统显示,信达生物最早于2016年1月申请信迪利单抗临床试验,2016年9月初获批试验许可。远远在7.22临床试验数据核查之后,最初申报的临床研究也非一线非鳞状非小细胞肺癌。最早申请非鳞状非小细胞肺癌是在2018年,彼时中国药物临床研究早已重新回归正轨。
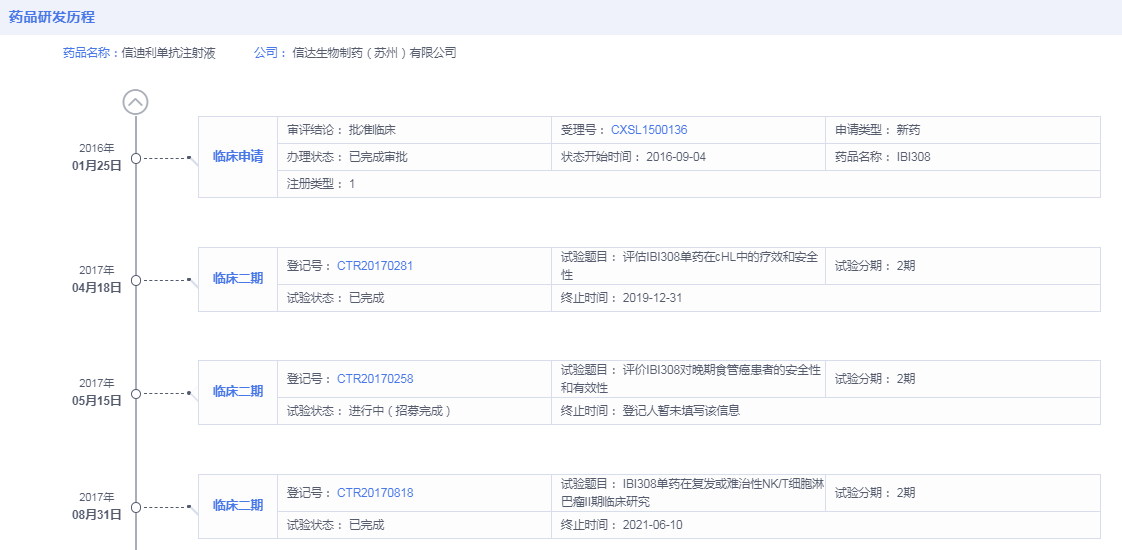
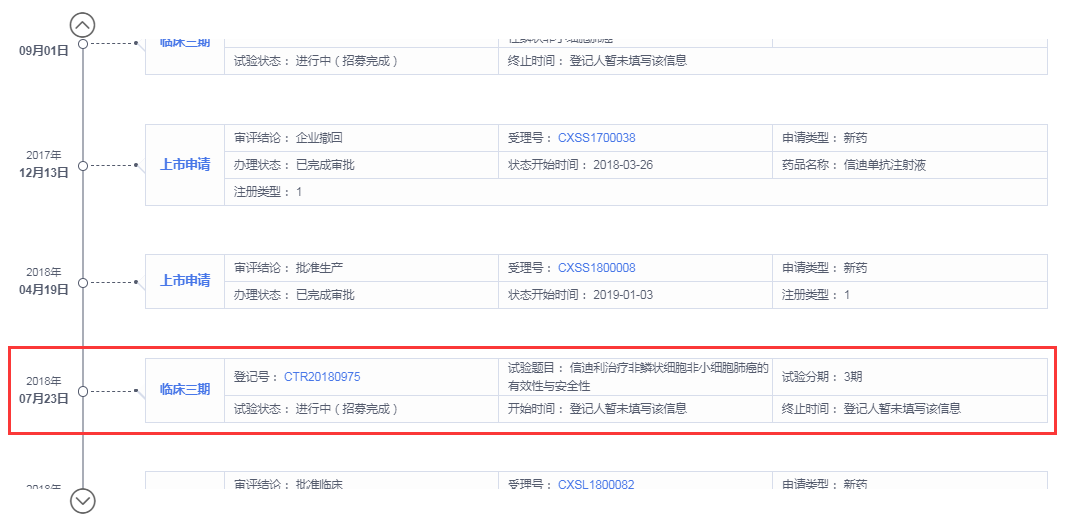
此外,本次审评会议,FDA实际也是认可信迪利单抗临床研究数据的。FDA对信达生物的48个临床中心进行调查,肯定了信达临床人员的专业。对信迪利单抗ORIENT-11临床研究结果也给予了肯定,同时表示对信迪利单抗没有任何对于安全性和有效性问题的质疑。主要矛盾争议点在于,FDA认为ORIENT-11在设计上显示缺乏多样性,需要补充研究信迪利单抗对于美国患者有效的研究。
事实上自7.22临床数据核查后,真实、规范、完整的临床试验一直是中国新药研发重点监管对象,中国新药临床研究已与国际接轨,数据不真实已几乎不存在。正如唯一支持信迪利单抗的南加州大学的JorgeNieva所言“经FDA充分检查,没有证据表明信达生物所提供的数据是不可靠的、合成的或其他欺诈性的。”故而,80%的中国临床研究存在欺诈或者不够标准不应该是被支持的数据证据。
结语
不过,我们应该正视历史,以此为经验教训,引以为戒。踏踏实实做研究,一步一个脚印,不能让黑历史成为我们药物创新前进路上的绊脚石。尽管这对于中国药物创新走向国家没有什么影响,但总会给予审评专家带来一种新的暗示,潜意识认为中国药物临床试验存在问题。
同时也希望无论是FDA、EMA抑或其他国家药品监管机构,不要每回有关审评中国药物临床数据时都将“黑历史”翻出来算旧账。
用发展的眼光看问题。
本文转自:药智网